
{kind=link}
You bent your thumb backward to your wrist at lunch. The whole table screamed. You did it again — because honestly, why wouldn’t you? It didn’t hurt. It was your thing.
Then there were the bruises. The massive, unexplained purple ones that mapped your shins like a bad watercolor. Your mom stopped asking how you got them around age nine.
And the pain. God, the pain. You used to have such charley horses in your legs that you would cry into your pillow at night. And every grown-up you knew told you the same thing: Growing pains. You‘re going to outgrow them.
You didn’t.
If you’re reading this and your chest just tightened — keep going. Because what you‘ve got to remember is it may not have actually been clumsiness and lack of strength or a bit of drama. It could have been Ehlers‑Danlos!. And nobody told you.
EDS is a group of 13 genetic disorders affecting connective tissue — the collagen-based scaffolding that holds your joints, skin, organs, and blood vessels in place. When that scaffolding is faulty, things stretch too far, tear too easily, and fall apart in ways that are invisible on X‑rays. The most common subtype is hypermobile EDS (hEDS), but its exact share of all EDS cases is uncertain and likely under‑recognized. Older estimates like “1 in 5,000” for EDS as a whole are now thought to underestimate how many people are actually affected.
This isn’t a diagnostic tool. Only a specialist can evaluate you for EDS. But if multiple items on this list feel like someone pulled pages from your childhood diary — that’s worth a real conversation with a geneticist or rheumatologist.
What you’ll find here:
- Twenty-three signs grouped by body system and lived experience — not just a random list
- Why these symptoms get missed for years (sometimes decades)
- A concrete step-by-step plan for what to do if this resonates
- How EDS is actually diagnosed — and what the process looks like
- Six common questions answered without the usual runaround
Table of Contents
What Is Ehlers-Danlos Syndrome — and How Does an Entire Childhood of Symptoms Get Missed?
The Short Version
Connective tissue is everywhere in your body. Joints. Skin. Gut lining. Blood vessel walls. When the genes responsible for building that tissue carry mutations — which is what happens in EDS — the structural integrity of all those systems gets compromised. Not all at once. Not always obviously. But persistently, and from birth.
There are 13 subtypes. Most people reading this are thinking about hypermobile EDS, which is by far the most prevalent. But here’s the frustrating part — there’s no genetic test for hEDS. Not yet. The gene or genes responsible haven’t been identified, despite a massive ongoing study (the HEDGE project) trying to find them. So diagnosis depends entirely on clinical criteria. Physical exam. Medical history. Pattern recognition. And a doctor who actually knows what to look for.
The Ehlers-Danlos Society has acknowledged that hEDS is likely far more common than previously estimated. When you factor in Hypermobility Spectrum Disorders — a related umbrella for people who have significant symptoms but don’t tick every diagnostic box — the number of affected individuals grows substantially.
So Why Did Nobody Catch It When You Were Seven?
A few reasons. And they stack.
Kids are bendy. That’s just… biology. A pediatrician sees a flexible child and thinks “normal.” Not “connective tissue disorder.” The threshold for concern is calibrated too high, especially for a condition most doctors learned nothing about in medical school.
Then there’s the silo problem — and this one drives patients absolutely nuts.
Your gastroenterologist focused on the stomach pain, the orthopedist on the ankle sprains, and the dermatologist on the odd scarring. But nobody sat in a room and said, “Wait. What if all of this is connected?” Because that’s not how our healthcare system is built. Specialists treat organs. EDS affects the glue between them.
And seriously? There is also the issue of gender; a large proportion of the diagnosed hEDS population are women, and in some clinic‑based surveys the female to male ratio is as high as 9:1. The reasons for this imbalance are still under discussion as to whether it is biological or diagnostic bias. What is known however is women‘s pain is not taken seriously and those who complain about it are told they are anxious while the males are given scans and blood tests to be referred to a specialist. This is well documented and upsetting.
Patient surveys from the UK and other countries suggest the median delay from first symptoms to an EDS diagnosis can be around a decade, and for some people it stretches into several decades.
23 Signs You Grew Up With Ehlers-Danlos Syndrome
Quick reality check before we start. Having two or three of these doesn’t mean you have EDS. Plenty of healthy kids are flexible or bruise easily. What matters — what actually gets a clinician’s attention — is the pattern. Multiple signs, across multiple body systems, over years.
One more thing. And this is serious. If you ever experience sudden severe chest pain, unexplained internal bleeding, or abdominal pain that feels catastrophic and comes out of nowhere — go to an emergency room. Vascular EDS is a rare subtype that can cause arterial rupture and organ perforation. Those symptoms don’t wait for an appointment.
Joints and Movement
1. Your “Party Tricks” Were Actually Joint Hypermobility
Bending your fingers flat against the back of your hand. Elbows that go past straight. Knees that bow backward when you lock them. You called them party tricks. Your friends thought they were gross or impressive or both.
What they actually were? Clinical signs of generalized joint hypermobility — the defining feature of hEDS.
Not every flexible person has EDS. That matters. About 1 in 30 people have some degree of hypermobility, and for most of them, it’s completely benign. The red flag is when flexibility comes packaged with pain, instability, weird skin, fatigue, and a trail of unexplained medical problems.
2. Joints That Popped, Clicked, and Slipped Out of Place
Knees cracking on the stairs. Shoulders grinding during a reach. Your jaw clicking every time you yawned. You thought everyone’s body sounded like that.
They don’t.
In EDS, subluxations — partial dislocations where a joint slides partway out of alignment and then back — happen during mundane activities. Opening a jar. Rolling over in sleep. Raising your hand in class. Some subluxations are painful. Others are so routine your body stops registering them. Full dislocations (joint completely out of socket) are less common in childhood but happen too — especially shoulders, kneecaps, and fingers.
3. Sprains and Injuries That Made No Sense
Twisted ankle on flat ground. Pulled muscle from picking up a textbook. Wrist that gave out during a cartwheel everyone else nailed.
This is what happens when connective tissue can’t stabilize your joints properly. You’re not doing anything riskier than the kid next to you. But your joints are working with weaker structural support — and nobody’s factored that in. So you get hurt more often, heal more slowly, and collect a reputation for being fragile. Or dramatic. Or both.
4. “Growing Pains” That Never Actually Stopped
The most universal EDS childhood memory. Hands down.
Deep, throbbing leg pain at night.
Pain that woke you at night, that your parents tried to soothe, that every pediatrician dismissed as “growing pains” before sending you home without real follow-up. Here’s the thing — real growing pains typically resolve by age 12. If your version persisted through your teens, required regular pain medication, or was severe enough to keep you out of school — that’s not a standard developmental pattern. That’s chronic musculoskeletal pain. And in the context of EDS, it often starts years before anyone thinks to check for a connective tissue problem.
5. You Were the “Clumsy Kid”
Tripping on nothing. Walking into doorframes. Dropping your phone, your fork, your dignity on a weekly basis.
But you weren’t careless. The actual issue — and this is something most people with EDS don’t learn until they’re diagnosed — is proprioception. That’s your brain’s sense of where your body parts are in space. When ligaments are lax and joints don’t send reliable position signals, your nervous system is essentially flying blind. Your coordination suffers not because you’re clumsy, but because your body is feeding your brain bad data. Huge difference.
6. PE Class Was a Nightmare
Some kids sat out. Others pushed through and ended up hurt. Plenty bounced between the two—a miserable cycle of trying, getting injured, and sitting on the sidelines in embarrassment.
Sports that involve repetitive impact (running, basketball, volleyball) tend to be rough on EDS joints. But here’s the paradox. Some kids with EDS gravitate toward activities that reward flexibility — gymnastics, dance, cheerleading, yoga. They’re genuinely good at these things, at first, because hypermobility is an advantage… until the injuries accumulate. The collision between “my body is great at this” and “my body keeps breaking” is confusing for anyone. For a ten-year-old, it’s isolating.
Skin and Healing
7. Bruises That Appeared Out of Nowhere
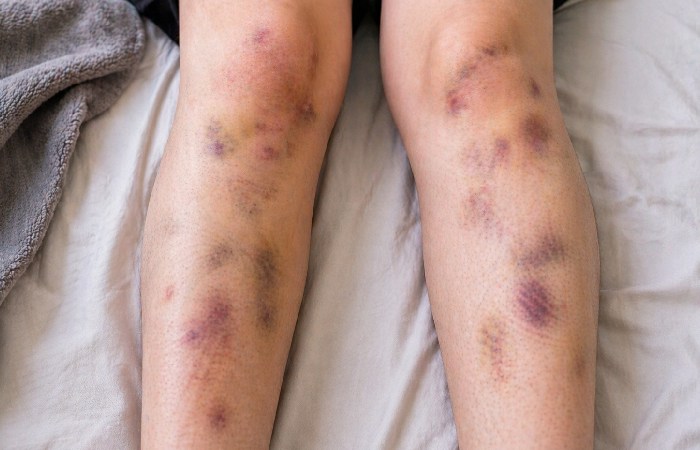
Monday morning. A bruise the size of a tangerine on your shin. Zero memory of how it got there.
In EDS, fragile connective tissue means fragile blood vessel walls. Minor bumps — the kind your body should absorb without a trace — rupture capillaries and leave visible marks. Some kids bruise so easily and so frequently that it raises questions — uncomfortable ones — about their safety at home.
If your legs were a permanent gallery of yellow and purple splotches that nobody could explain, and it started before you were old enough to be playing contact sports — yeah. That’s a pattern worth noting.
8. Skin That Felt Different
Unusually flexible. Others would say ‘soft’. People would notice if your skin was soft – perhaps if it stretched farther than your friends’ skin when you pulled on it (everyone with EDS tries this), or if you could see your veins through it.
The skin is different for the subtypes. Classic EDS usually has very hyperextensible fragile skin with broad atrophic scars. Hypermobile EDS often has softer skin, which is sometimes mildly stretchy and has a tendency to bruise easily. Vascular EDS can have transparent, thin skin over the chest and abdomen which is the presentation which may suggest blood vessel fragility.
9. Cuts That Took Forever to Heal
A scraped knee that stayed open for weeks. Paper cuts that kept reopening. Scars that widened instead of fading — thin, shiny, sometimes described as tissue-paper texture.
Collagen drives wound repair. When collagen is structurally defective, healing slows down and the results look different. Wide, atrophic (sunken) scars — especially on the knees, shins, elbows, and forehead — are a recognized clinical marker across several EDS subtypes. If your childhood knees looked like a road map of weird scars while your siblings healed cleanly from the same kinds of falls… that’s data.
10. Stretch Marks Before Anyone Else Had Them
Stretch marks at 13. On your thighs, hips, back, inner arms.
Your stretch marks showed up early—before any major weight changes, before pregnancy, long before anyone else your age was getting them. Your dermatologist probably shrugged. Called them striae. Said they were common. And they are common — in the general population. But early onset, unusual distribution (back, inner arms), and appearance without an obvious trigger suggest something structural. In EDS, skin proteins can’t handle normal growth-related stretching. The tissue splits because it lacks the elasticity to accommodate even typical adolescent changes. This sign gets missed constantly because stretch marks are so normalized that nobody investigates further.
Energy, Nerves, and Autonomic Function
11. Bone-Deep Fatigue That Rest Couldn’t Fix
Ten hours of sleep. Still exhausted. Couldn’t keep up on a hike. Battery at zero by 2 PM with no explanation and no amount of caffeine making a dent.
Where does this come from? Multiple places, actually.
Unstable joints force your muscles into constant overtime — they’re acting as substitute ligaments, holding your skeleton together, and that’s physically draining in a way that doesn’t show up on blood tests. Autonomic dysfunction (more on that next) disrupts sleep quality. Chronic pain is itself exhausting. And some research suggests mitochondrial function may be impaired in certain EDS patients — though that area is still under active investigation and hasn’t changed clinical protocols yet.
This isn’t laziness. It never was. It’s a body running a 24-hour stabilization marathon that nobody else can see.
12. Getting Dizzy or Blacking Out When You Stood Up

You get up from the couch The room turns grey. Darkness closes in. Your vision fades to tunnel vision. You may have grabbed the wall, or maybe you hit the floor. And someone says, “You stood up too fast.”
No. Your autonomic nervous system wasn’t functioning properly. Dizziness, greyout, or even near-fainting upon standing is a hallmark presentation of postural orthostatic tachycardia syndrome (POTS) and other forms of orthostatic intolerance. Many large clinics are now finding autonomic dysfunction to be quite prevalent in those with EDS and hypermobility related disorders, and POTS is one of the more frequently seen comorbidities, particularly amongst teens and young adults. EDS doesn’t just affect the connective tissue and skin, but also the blood vessels and autonomic nervous system that help govern blood pressure and heart rate. POTS and other forms of dysautonomia are very common in those with hypermobility related syndromes, causing dizziness, fast heart rate, and other symptoms upon standing. As a kid, that may have been dismissed as “drama,” but it’s real physiology.
13. Headaches and Migraines Starting Young
Headaches in elementary school that never fully went away. Tension-type. Migraines with aura. Light sensitivity that made fluorescent classrooms unbearable.
Multiple mechanisms drive this in EDS. Cervical instability — neck joints that won’t stay aligned — creates tension patterns that refer pain upward. Muscles overcompensating for loose ligaments tighten and spasm. TMJ dysfunction (jaw problems — we’ll get there) radiates into the temples. And autonomic dysregulation can trigger migraine-type episodes on its own.
14. Your Body Couldn’t Regulate Temperature
Always too hot in summer. Always too cold in winter. Sweating through a t-shirt during mild activity. Or barely sweating at all while everyone else dripped.
Temperature regulation is autonomic. When that system is dysregulated — as it frequently is in EDS — your body’s thermostat goes haywire. You’re not “just sensitive.” You’re not dehydrated (although, fine, you probably should drink more water). Your nervous system is genuinely struggling with a basic regulatory function. It’s one of those symptoms that gets dismissed so casually — “just wear a sweater” — that most people stop mentioning it entirely.
15. Brain Fog Before You Knew the Term Existed
Losing your train of thought mid-sentence. Reading the same paragraph four times and absorbing nothing. Walking into a room and standing there, blank, at age twelve.
“Brain fog” isn’t an official diagnosis. That’s part of the problem — there’s no billing code for it, so it’s easy for clinicians to dismiss. But cognitive dysfunction is reported so widely and so consistently in the EDS community that ignoring it isn’t credible. The likely drivers? Reduced cerebral blood flow from autonomic dysfunction. Fragmented sleep. Chronic pain consuming cognitive bandwidth. And possibly — this is newer research — mast cell-mediated neuroinflammation.
It’s hard to pay attention in school when your brain is literally not getting enough blood every time you sit upright for forty minutes. Nobody factors that in when they’re evaluating your grades.
Gut, Bladder, and Systemic Clues
16. Stomach Problems That Stumped Every Doctor
Nausea after meals. Bloating that made you look pregnant by dinner. Alternating between not going to the bathroom for days and then… the opposite. Maybe someone wrote “IBS” in your chart and moved on.
Connective tissue lines the entire GI tract. All of it. When that tissue is compromised — as it is in EDS — motility goes sideways. Food moves too slowly (gastroparesis). Acid goes where it shouldn’t (reflux). The gut wall itself may be more permeable than it should be. None of this is visible on a standard scope. So the standard workup comes back “normal,” and you get told to eat more fiber.
A kid who can’t eat without stomach pain isn’t being picky. A teenager who bloats after every meal isn’t overreacting. But good luck getting taken seriously at fifteen when your labs look fine and the gastroenterologist has already mentally moved on to the next patient.
17. Allergies, Hives, and Reactions to Everything
You were the kid who was allergic to everything. Exercise gave you hives. Heat made you flush crimson. Random foods triggered cramps — and the list kept changing. Or perhaps you always found yourself carrying an EpiPen without anyone ever bothering to tell you exactly what you were allergic to because the allergy testing was showing up pretty much nothing.
Many patients and a number of the practitioners refer to this triad, the triad of EDS or hypermobility, POTS and mastcell–mediated symptoms such as MCAS since it is so commonly encountered in clinics and support groups. While the research is always developing, if you suffered from chronic hives, wheezing or flushing or other cystitis symptoms, or food reactions without cause along with your joint issues, this pattern is worth bringing to your doctor‘s attention. As a child, it may have been represented by your chronic hives, unexplained rashes, inexplicable food allergies or drug reactions with no “culprit. “A pain to manage. A pain to explain.
18. Bladder Issues Nobody Talked About
This one gets skipped in most lists. For obvious reasons — nobody wants to discuss it.
But pelvic floor dysfunction is recognized as a common feature of EDS, and it often starts early. Frequent urination. Sudden urgency. Occasional accidents at an age when that’s socially devastating. The pelvic floor is made of connective tissue and muscle. When the tissue is lax — which it is in EDS — the structures it supports don’t function properly.
If you had unexplained bladder issues as a kid or teenager and felt ashamed to bring them up — you’re not alone, and it’s worth mentioning to a specialist now. Even if it feels like ancient history.
19. Dental Crowding and a Jaw That Clicked
High-arched palate. Teeth crammed together no matter how many rounds of braces you endured. A jaw that clicked, locked, popped, or ached when you chewed.
Your jaw is a joint. Connective tissue holds it together. So it’s susceptible to the same instability as every other joint in your body. TMJ dysfunction in EDS patients is common — and it’s annoying because it affects eating, talking, yawning, and sleeping.
And then there’s the orthodontic merry-go-round. Braces straighten the teeth. You get the braces off. The teeth shift right back. Because the connective tissue and bone structure underlying the alignment isn’t stable enough to hold the correction. Your orthodontist was probably confused. You were probably frustrated.
One more thing about dental visits — and people in the EDS community bring this up constantly. Local anesthetic resistance. Standard doses of lidocaine don’t fully numb the area.
The anesthetic wears thin. The drill bites, the stitches tug, and the pain cuts through—so you speak up. And the dentist looks at you like you’re making it up.
Standardized clinical practice guidelines are still behind where the patient reports have been for years. However, awareness is spreading, and increasing numbers of anesthetists with some knowledge of EDS are adapting their protocols to account for this.
School, Identity, and the Emotional Toll
20. Being Called Lazy, Dramatic, or a Hypochondriac
This one cuts deep.
Missing school for pain no test could prove, you kept asking to sit out of activities because your body hurt in ways no scan could explain. They visited the nurse‘s office so often that after a while they knew your name and then some of them began to wonder if it was even real.
And at some point along the way, the cover story changed from “things may be wrong” to “you are wrong”.
You‘re only trying to leave class. Other children don‘t complain that way. There’s nothing wrong — your tests are normal.
Research published through the National Institutes of Health has documented that hEDS patients are frequently misdiagnosed with somatic symptom disorder or told their pain is psychogenic. The clinical term for what many patients experience is medical gaslighting — and it’s not a fringe complaint. It’s systemic.
Being told your pain isn’t real doesn’t make it disappear. It teaches you to stop asking for help. That’s a different kind of damage, and it compounds over years.
21. Anxiety and Depression That Weren’t “Just” Mental Health
Here’s where things get tangled.
Were you anxious about the fact you were carrying an undiagnosed long term illness and no one believed you? Or were you anxious as a co-morbid condition overlapping biologically? Probably some of both.
A 2025 genome-wide association study — a large-scale meta-analysis of hEDS — found genetic correlations between hEDS and depression, anxiety, and autism spectrum disorder. So the link isn’t purely reactive. There appear to be shared biological pathways.
And then there is the POTS layer. POTS can give you tachycardia, tightness in the chest and adrenaline surges just from standing up. Those feelings are exactly the same as with a panic attack. If you spent your teens on SSRIs for “anxiety” while the real driver was undiagnosed autonomic dysfunction… that’s a frustrating realization to arrive at in your thirties.
None of this means the anxiety isn’t real. It is. But treating only the psychological symptoms while ignoring the physiological fuel is like mopping the floor with the faucet running. Wet floor forever.
22. Struggling With Handwriting and Fine Motor Tasks
Messy handwriting. Painful pencil grip. Buttons and zippers taking twice as long as they should. Maybe you got an OT evaluation. Maybe everyone just decided you weren’t trying.
The tiny joints of the hand — the fingers, thumb, and wrist — are some of the most common sites to be affected in hEDS. The joint instability in these fingers means being able to write for any length of time leaves you in pain and exhausted. With proprioceptive direction issues (your brain isn’t sure where your fingers are) and compromised grip strength, suddenly any writing task is a burden.
For school-age children, this is academically brutal. Your hands hurt when you write. Nobody understands why. So you either push through the pain — which burns you out — or you fall behind. Neither option is acceptable. And both are preventable with the right diagnosis and accommodations. Typing instead of handwriting. Modified grip tools. Extended test time. These aren’t luxuries. They’re access.
23. The Constant Feeling of Being Different
No test measures this. No scan picks it up. But it’s the sign that ties all the others together.
You were always in this weird lag with everyone else. Other children could run, and play, and eat anything they pleased without anything happening afterward. You weren‘t; you were exhausted while everyone else was energetic, sore when everyone else was fine, laden with a silent weight that no one could see or comprehend.
And the worst part — you started believing what they said about you. That you were weak. Sensitive. Broken.
Getting a name for it doesn’t erase those years. But for a lot of people, it’s the first step toward understanding that the problem was never you. Your body was operating with different hardware. Nobody handed you the manual. And that’s a failure of the system — not a flaw in who you are.
Symptoms That Fly Under the Radar
The 23 signs above cover the patterns most commonly recognized. But EDS has a few features that almost never make the standard listicle. If you’ve nodded along to several signs already, these might resonate too.
- ADHD connection. There’s a statistically significant link between joint hypermobility and ADHD. The mechanism isn’t fully understood — it might be comorbid, it might share neurological roots, it might be something else entirely. What’s clear is that a lot of kids with undiagnosed EDS get an ADHD diagnosis first. Especially boys. If your childhood ADHD treatment didn‘t seem to work quite right, connective tissue issues may be the diagnostic unknown.
- Hormonal shifts at puberty. Many women with hEDS report that everything got worse around age 13. Symptoms that were manageable before puberty suddenly intensified — more pain, more fatigue, more instability, more GI issues. The influence estrogen has on connective tissue laxity is direct, however variability in hormone levels through a menstrual cycle can influence the severity of EDS symptoms monthly. If you had a reasonable childhood but found your teenage years to be a “hit”, this hormonal connection may be relevant.
- Heavier, more painful periods. Connective tissue acts as the support between the pelvic organs. That support is usually terrible in EDS and as such it as been linked to menorrhagia, dysmenorrhoea and endometriosis-syndromes. This aspect is so significantly underserved in most EDS contentits impact on such a large portion of the patient population often not being addressed with anywhere near the amount that it needs treating.
Why It Takes So Damn Long to Get Diagnosed
The average diagnostic delay for hEDS exceeds a decade. That’s not a minor gap. That’s a generation of childhood spent without answers.
Why is there no easy diagnostic test for our most common type? Because it hasn’t been found yet. There is no blood test, biopsy, or imaging study that can say a clear yes or no. The 2017 international diagnostic criteria for HEDS require a skilled clinician to do a physical exam, take a history and work within a framework. Most primary care doctors have never seen those criteria. Many don’t know they exist.
Imagine a woman in her late twenties. She‘s waiting in the office of yet another rheumatologist. She‘s been to four this year; the first told her she had IBS, the second told her she was anxious, and the third told her her loose joints were no big deal. No one ever looked at the whole picture. And this isn‘t an imaginary case, it‘s just the average EDS patient experience in our healthcare system. And this has been recorded in the medical literature.
The list of clinical symptoms for EDS (and hEDS in particular) is such that they are often confused with other conditions such as fibromyalgia, chronic fatigue syndrome, lupus and anxiety disorders. People with hEDS are often wrongly diagnosed with ‘something else’, often going around in circles for three, four or five diagnoses until someone puts the connective tissue pattern together.
Years pass before that happens. By then, damage has already been done—and any remaining faith in the healthcare system is gone. Then there’s the “you’ll grow out of it” problem. Most kids with hypermobility are fine. Pediatric flexibility is common and usually benign. So clinicians are trained to reassure. Which means the rare child who actually has EDS gets swept into the “probably fine” pile alongside everyone else. Nobody’s wrong for reassuring — but the default assumption fails a small, identifiable subset of kids who needed investigation, not reassurance.
What to Do if These Signs Sound Like Your Childhood
Reading a list and thinking that’s me isn’t the same as having a diagnosis. But recognition is the first step. Here’s how to move from “I think this might be what I have” to actually getting evaluated.
Step 1 — Write Down What You Remember
Go back as far as you can. Document:
- Joint issues a. Dislocations, subluxations b. Chronic pain c. Specific site
- Skin things (how often one bruises, scars look, when one gets stretch marks)
- Energy on a scale of 0 to 10, 0 when fatigues started, how bad they were, and how it stopped you from doing whatever you were doing.
- Consequences of gut e.g. Bloating, constipation, diarrhoea, food reactions, reflux.
- Autonomic weirdness – lightheadedness when you stand, fast heartbeat, trouble with body temp regulation
- Family history and this one is significant. Do your folks or sibs have the same symptoms? History of connective tissue abnormality, aneurisms, or anything else along these lines?
Photos help. Old photos of hypermobile positions, unusual scars, or bruising patterns are genuinely useful clinical evidence. Dig through your phone, your parents’ photo albums — whatever you’ve got.
Step 2 — Look Up the Beighton Score (But Don’t Diagnose Yourself)
Beighton Score Assessment of joint hypermobility. Five sites: fifth finger, thumbs, elbows, knees, trunk flexion. Eight possible points. Adults less than 50: 5 or more points- hypermobility in the new (2017) diagnostic criteria:
You can look up the maneuvers and get a rough idea of where you stand. That’s fine. Useful, even. But the Beighton Score is one component of a larger diagnostic framework — not a standalone answer. Meeting the threshold doesn’t confirm EDS. Falling short doesn’t rule it out (especially if your joints have stiffened with age or injury). A specialist interprets the score in context. That’s their job.
Step 3 — Find a Specialist Who Actually Knows EDS
This is the hard part. Not every doctor is equipped for this evaluation.
- Geneticists — best for confirming or ruling out rarer subtypes through genetic testing
- Rheumatologists particularly rheumatologists experienced with connective tissue disease (not all rheumatologists are EDS-literate…ask before booking).
- Physiatrists: Are PM&R doctors who specialize in musculoskeletal and functional problems
- EDS-aware physical therapists — who understand hypermobility-specific rehab
The Ehlers-Danlos Society has a provider directory that can help. Wait times for specialized clinics can be long — months, sometimes — so get your name on a list early.
Step 4 — Show Up Prepared
Bring your symptom timeline from Step 1, relevant medical records — imaging, past diagnoses, specialist notes, and a medication history. Bring photos if you have them, and a short family medical history.
And if you’ve been dismissed before — say so. Calmly, directly. What if was more like “I‘m told this is anxiety before, but I‘m interested in finding out if a connective tissue problem can account for the pattern as a whole” noting the proposed issue allows room for investigation without putting her on the defense. It’s not a guarantee — but it shifts the conversation.
How Diagnosis Actually Works
The process is clinical. There is no lab, scan, or single definitive test that can give a simple yes or no. An expert uses the international set of 2017 definitions, which depend on three conditions being met at the same time:
- One. True generalized joint hypermobility verified by application of the Beighton Score taking into account age references.
- Two. Two (minimum) systemic features from a set checklist examples include skin, dental/palatal, family history, musculoskeletal, mouth, eye, other, other systemic complications and other.
- Three. Diagnostic possibilities eliminated most notably Marfan syndrome, osteogenesis imperfecta, and the types of EDS with established genetic etiology.
For classical EDS, vascular EDS, and other rarer types, genetic testing can confirm the diagnosis definitively. If a test identifies a mutation in COL5A1 (classical) or COL3A1 (vascular), that’s conclusive. If the test is negative, it doesn’t rule out hEDS — because nobody’s found the responsible gene yet. That’s not a flaw in your case. It’s a gap in current science.
The HEDGE study — the largest genetic research project focused on hEDS — is working to close that gap. When it does, diagnosis will likely become simpler and more accessible. But right now, we work with what we have.
Living With EDS After a Late Diagnosis
Diagnosis as an adult — after years of “nothing’s wrong” — produces a complicated cocktail of feelings. Relief. Grief. Anger. And sometimes a weird guilt that maybe you’re still making too much of it.
You’re not.
But diagnosis also isn’t a cure. It’s a starting point. And the starting point matters because it redirects everything.
- Physical therapy — but the right kind. Standard PT can actually make EDS worse if the therapist pushes hypermobile joints into deeper ranges. What you need is someone who understands that “more flexible” isn’t the goal. Stabilization is. Proprioceptive training. Gradual, joint-protective strengthening. If your PT tells you to stretch more, find a different PT.
- Pacing is a survival skill. Learning to distribute your energy across a day — instead of crashing through boom-and-bust cycles — is one of the highest-impact changes EDS patients make. It feels frustrating. It looks like “doing less.” But the alternative is flaring for three days after one good day, and that math doesn’t work.
- This is not an optional mental health service. After years of gaslighting, self-doubt and invisible suffering are scars. Finding a therapist who understands chronic illness (preferably one who is trauma-informed and won‘t holus-bolus try to fix you with breathing exercises) is worth the search. Trauma-informed techniques such as ACT or somatic experiencing tend to be more effective than bland talk therapy.
- Community helps more than you’d expect. The EDS community — through the Ehlers-Danlos Society, Reddit, Instagram, local support groups — provides something clinical care can’t. Recognition. The experience of telling someone a symptom and watching them nod because they have it too. That’s not a small thing. There are few greater reliefs than being totally understood by a total stranger on the internet after years of being told you are just imagining it.
Frequently Asked Questions
Q: Can you have EDS and not know until you’re an adult?
More common than anyone should be comfortable with. Median diagnostic delay is over 10 years. Plenty of people get diagnosed in their thirties or forties — after spending their entire childhoods being told they were fine, anxious, or making it up. The system failed them. Full stop.
Q: Is being double-jointed the same as having EDS?
No. Being double-jointed — hypermobile — is common. About 1 in 30 people. Most of them are perfectly healthy. EDS is hypermobility plus a constellation of other symptoms — skin issues, chronic pain, autonomic problems, GI dysfunction, and more. One piece of a puzzle isn’t the whole picture.
Q: At what age do symptoms usually start?
Most people with hEDS can trace things back to early childhood. The pain, the flexibility, the bruising, the fatigue — it was all there. But the 2017 diagnostic criteria were designed with adults in mind, and some features — like certain skin changes — may not become obvious until adolescence or later. A pediatric-specific diagnostic framework is being developed. It’s overdue.
Q: Can kids with EDS play sports?
It depends. Sport such as swimming, cycling and lower-impact strength work should be safe and helpful (even if they still carry some risk). Contact sports and higher-impact sports (running, basketball) are more risky and should probably be modified. You‘re not trying to wrap your kid in bubble-wrap you‘re trying to include them with knowledge and a safety net. A knowledgeable sports-enthusiast-friendly PT who understands EDS may be able to create something realistic.
Q: Which doctor diagnoses EDS?
Geneticists and rheumatologists with connective tissue experience are your best bet. But — and this is the frustrating part — not all of them are EDS-literate, and many have long wait lists. The Ehlers-Danlos Society’s provider directory is a reasonable starting point. You may need to advocate for the referral. That shouldn’t be necessary. It often is.
Q: Does it run in families?
Yes. Most EDS types are inherited in an autosomal dominant way, where one altered gene from one parent is enough and each child has about a 50% chance of inheriting it. Some rarer subtypes are autosomal recessive and require changes from both parents. What often happens is that one person gets diagnosed, and then everyone starts looking around the Thanksgiving table going, “Wait… Mom, do your thumbs do that too?” Recognition cascades through families. It’s one of the stranger and more clarifying experiences of adult life.
Medical disclaimer: This article is presented for educational purposes only and is not a diagnostic or treatment tool. It cannot determine if you do or don‘t have EDS or any associated disease/disorder. Please speak to a medical professional if you are concerned about your symptoms or treatment.
About the Author:
Abdul Rahman, has more than 4 years experience writing about consumer electronics, laptops and IT support solutions in Ireland and the UK. He simplifies complicated repair terms into easy, useful advice so you can be sure of your buying decisions.
Published by: www.theglamourmedia.com a convenient source of content on business, health, technology and lifestyle that strives for relevance and use rather than sophisticated implementations and complex concepts.